Smith Lemli Opitz sindrom je kongenitalni razvojni poremećaj koji se, među ostalim manifestacijama, karakterizira prepoznatljivim crtama lica, intelektualnim teškoćama i smetnjama u učenju, problemima u ponašanju i malom glavom (mikrocefalija). Uz malformacije važnih organa poput bubrega, srca, genitalija i crijevnog trakta, djeca s ovim stanjem pokazuju i karakteristike autizma i poremećaja hiperaktivnosti s nedostatkom pažnje (ADHD).Većina oboljelih imaju spojeni drugi i treći nožni prst, a neki mogu imati i dodatne prste. Stanje je relativno rijetko, pogađa približno jedno na svakih 20 000 do 60 000 novorođenčadi.
SeventyFour / Getty ImagesSimptomi
Znakovi sindroma Smith Lemli Opitz prisutni su pri rođenju, a njihova težina jako varira. U 80 posto do 99 posto ovih slučajeva. vide se ove karakteristike:
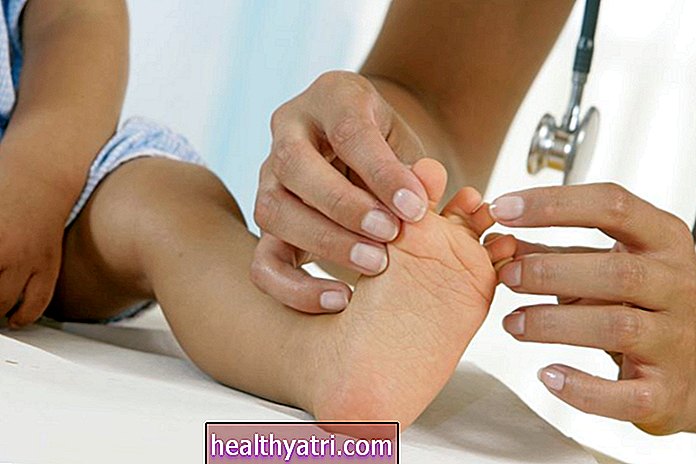
- Prepleteni nožni prsti: Uobičajena značajka stanja je spajanje između drugog i trećeg prsta, stanje koje se naziva „sindaktil“.
- Intelektualni invaliditet: Iako se stupanj toga može razlikovati, ovo stanje često dovodi do poteškoća u učenju.
- Nenormalno mala lubanja: Još jedna od značajki manja od prosječne veličine lubanje, stanje koje se naziva mikrocefalija.
- Abnormalne crte lica: Oni sa Smith Lemli Opitz sindromom imaju karakteristične crte lica, uključujući manju donju čeljust i širok, ravan nos. U rjeđim slučajevima osobe mogu imati obješene kapke, mačje oči, male ili odsutne oči, kao i široka usta.
- Teškoće s hranjenjem: U dojenčadi ovo stanje može dovesti do poteškoća s dojenjem, što utječe na razvoj.
- Donji mišićni tonus: Uobičajena karakteristika sindroma je niži mišićni tonus.
Brojni su rjeđi simptomi koji se javljaju u bilo kojem od 5 do 79 posto slučajeva, uključujući:
- Anomalije razvoja zuba: Rano nicanje odraslih zuba i povećane desni znakovi su sindroma Smith Lemli Opitz.
- Dvosmislene genitalije: Genitalije zahvaćenih mogu biti manje definirane. Muškarci će to vjerojatnije doživjeti s nedovoljno razvijenim penisom i nespuštenim testisima.
- Poremećaj hiperaktivnosti s nedostatkom pažnje (ADHD): Ovaj razvojni poremećaj karakteriziraju poteškoće u regulaciji ponašanja i impulsa, kao i hiperaktivnost.
- Autizam: Također poznato kao poremećaj iz autističnog spektra (ASD), ovo stanje dovodi do oštećenja socijalnih vještina, govora i neverbalnih komunikacijskih sposobnosti, kao i ponavljajućeg ponašanja.
- Srčane mane: Srčane mane povezane sa Smith Lemli Opitzovim sindromom uključuju razvoj rupe u zidu između dvije gornje komore (defekt pretkomorske pregrade) ili one između donjih komora (defekt ventrikularne pregrade).
- Promijenjena anatomija šake: Osobe s tim stanjem mogu imati suviše male prste na rukama i nogama. Osim toga, položaj palca može biti i netipičan po tome što je bliže zglobu. Prijavljeni su i umreženi prsti. Također je zabilježena ruka kandže, atipična zakrivljenost prstiju.
- Fotosenzibilnost: U mnogim je slučajevima koža zahvaćenih posebno osjetljiva na sunčevu svjetlost.
- Česta infekcija: Osobe sa sindromom imaju povišen rizik od bakterijske infekcije.
- Rascijepljeni jezik: U otprilike pet do 30 posto slučajeva zahvaćeni će imati rascijepljeni jezik u kojem je vrh podijeljen.
- Abnormalnosti kralježnice: Uz ostale deformacije kralješaka, skolioza - bočna zakrivljenost kralježnice - kao i kifoza ili grbav, također mogu pratiti stanje.
- Napadaji: Osobe s ovim stanjem sklonije su razvoju napadaja.
- Nehotični pokreti oka: nekontrolirani i brzi pokreti oka (nistagmus) također mogu pratiti sindrom.
Uzroci
Smith Lemli Opitz sindrom genetski je poremećaj uzrokovan mutacijom gena DHCR7. Ovaj gen regulira važan enzim, 7-dehidrokolesterol reduktazu, koji je uključen u proizvodnju tjelesnog kolesterola. Među svojim funkcijama, kolesterol je glavna komponenta staničnih membrana i pomaže u stvaranju mijelina, tvari koja štiti moždane stanice (neurone). Također igra značajnu ulogu u pravilnoj probavi.
Mutacija DHCR7 uzrokuje nedostatak 7-dehidrokolesterol reduktaze uzrokujući deficite u proizvodnji kolesterola. Također omogućuje da se otrovni nusproizvodi kolesterola nakupljaju u tijelu, što ometa razvoj i rast u više tjelesnih sustava. Točan mehanizam kako ovaj nedostatak kolesterola dovodi do sindroma Smith Lemli Opitz još uvijek se istražuje.
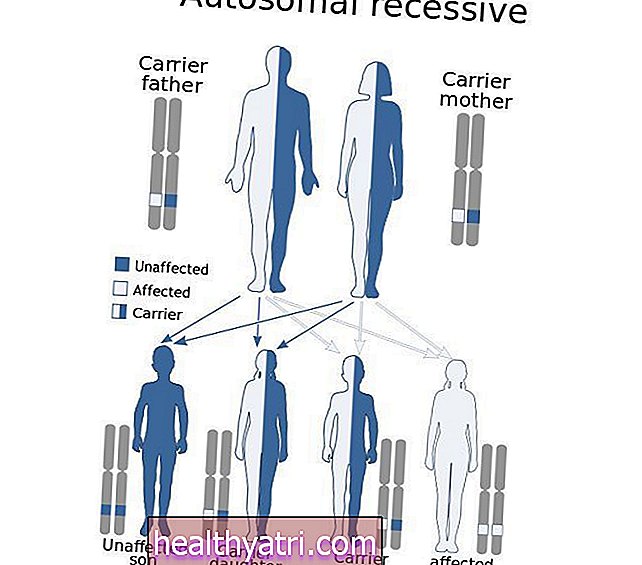
Genetski nedostatak, ovo stanje slijedi ono što se naziva "autosomno recesivni obrazac", što znači da su mu potrebne dvije kopije gena - po jedna od svakog roditelja. To znači da roditelji oboljelih nose gen, ali ne moraju i sami imati simptome.
Dijagnoza
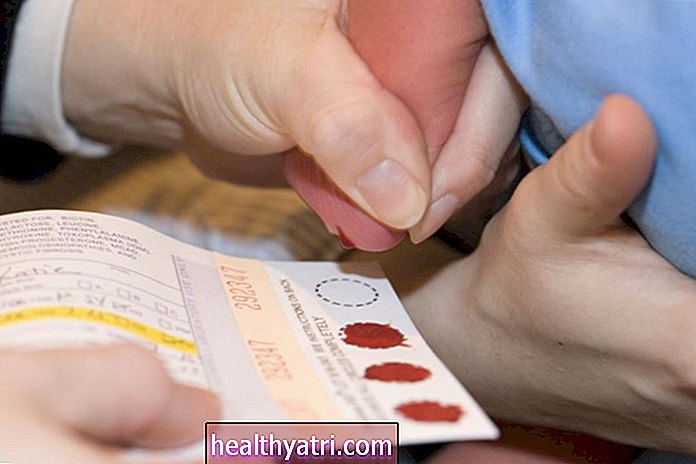
Kao i kod drugih urođenih bolesti, dijagnoza Smith Lemli Opitza uključuje procjenu fizičkih simptoma, kao i ispitivanje omjera 7-dehidrokolesterol reduktaze i kolesterola, što se radi pomoću krvnih pretraga za sumnjive slučajeve. Uz to, prenatalno genetsko testiranje također može otkriti mutacije gena DHCR7 koje dovode do razvoja stanja.
Liječenje
Preuzimanje ovog stanja uključuje koordinirani napor; budući da za ovo stanje ne postoji izravni lijek, simptomima i manifestacijama treba učinkovito upravljati. Takvi pristupi uključuju:
- Dodatak kolesterola: Iako je potrebno više istraživanja kako bi se procijenila učinkovitost ovog pristupa, prehrana bogata kolesterolom - uz uzimanje dodataka - može pomoći u smanjenju nekih simptoma.
- Fizikalna terapija: Pristupi fizikalnoj i radnoj terapiji, ako se daju pravovremeno, mogu pomoći kod invaliditeta povezanih s tim stanjem.
- Medicinski tretmani: Dostupni su pristupi za preuzimanje nekih fizičkih simptoma sindroma Smith Lemli Opitz, uključujući probavne poteškoće, probleme s vidom, kao i deformacije lica i drugih.
- Nadzor: Uspješno liječenje ovog stanja zahtijeva dosljedno praćenje fizičkih simptoma, zastoja u razvoju i prehrambenih čimbenika.
Prognoza
Dobra vijest je da, ako se pravilno upravlja sindromom Smith Lemli Opitz i pruži im odgovarajuća medicinska pomoć, oni koji imaju to stanje imaju potencijal da imaju normalan životni vijek.To je rečeno, samostalan život nije vjerojatan zbog teške intelektualne invalidnosti često prati ovaj sindrom. Značajno je da je preživljenje novorođenčadi s ozbiljnim simptomima ozbiljno oštećeno i postoji vjerojatnost smrti u roku od nekoliko mjeseci.
Snalaženje
Veliki urođeni poremećaj poput sindroma Smith Lemli Opitz predstavlja značajan izazov za pogođenu osobu, njenu obitelj i liječnike. Iako je moguće uspješno upravljanje, nema sumnje da postoji značajan psihološki pad od ovog tereta. Oni koji se stave u poziciju da se brinu za nekoga s ovom bolešću mogu smatrati korisnim savjetovališta ili grupe za podršku invalidima. Između ostalog, resurse poput veza do najnovijih službi za istraživanje i podršku okuplja Smith Lemli Opitz / RSH Foundation.
Riječ iz vrlo dobrog
Stanje koje je ovo iscrpljujuće i teško, koje može utjecati na toliko aspekata kvalitete života, može izgledati neodoljivo. Međutim, postojeći pristupi liječenju sindroma Smith Lemli Opitz ne samo da se neprestano usavršavaju i poboljšavaju, već su istraživanja ovog poremećaja u tijeku. Kako medicinska zajednica bude saznavala više o uzrocima i posljedicama ovog stanja - kao i o djelotvornosti pristupa liječenju - prognoza i kvaliteta života pogođenih samo će se poboljšati.
















.jpg)